Case Report

Primitive Neuroectodermal Tumor of Adrenal Gland
Vitorino Modesto dos Santos1,2, Marcos Correa da Trindade1, Viviane Vieira Passini Soares1, Victor Manabu Yano1, Mayza Lemes Duarte1
1Armed Forces Hospital and
2Catholic University of Brasília, Brasília-DF, Brazil.
"This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Submitted: Sunday, February 19, 2017
Accepted: Friday, April 21, 2017
Published: Sunday, April 23, 2017
Abstract
Primitive neuroectodermal tumor is a rare malignancy of the Ewing`s sarcoma family of neoplasms, constituted by small round malignant cells. They more often occur in children and young adults, and the prognosis is more favorable than in older adults. The occurrence of this tumor in adrenals is exceeding rare and case studies are scarce. A 27-year-old man is herein described with the adrenal tumor confirmed by immunohistochemistry and FISH methods.
Key words
Adrenal gland, diagnosis, primitive neuroectodermal tumor, young adult
Introduction
Primitive neuroectodermal tumors (PNETs) are rare cancers that belong to the family of Ewing's sarcomas and are characterized by small round cells [1-6]. They more often affect children and young adults, and the prognosis is less favorable among the older groups [2-4,6]. PNETs are classified as central type - if occur in the central nervous system (CNS); and peripheral type - if are located outside the CNS or in the sympathetic nervous system [2]. Primary adrenal PNETs are very uncommon, with few cases described worldwide [6-11]. Herein is reported the case study of a young male patient with an adrenal PNET, and the diagnosis was established by utilizing both immunohistochemical and FISH methods [6-11].
Case report
A previously healthy 27-year-old man had dry cough and burning epigastric pain during three months, and the upper gastrointestinal endoscopy revealed a gastritis and H. pylori positive. As there was no improvement with the specific treatment, the abdominal tomographic study was done and detected a large mass on the left adrenal gland that was excised (Figure 1A-C).
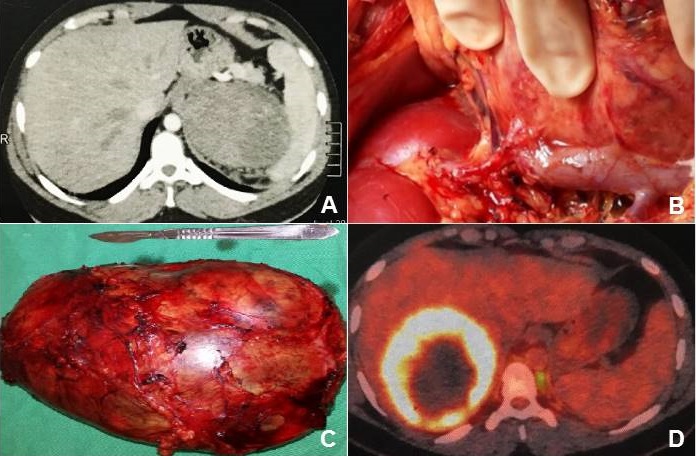
Figure 1:
(A) Abdominal tomographic image showing a conspicuous mass on the left
adrenal gland; (B) and (C): excision of the primary adrenal tumor,
which measured 18.5 x 16.0 x 12.5 cm; and (D): image of metastasis from
the adrenal PNET with 15 cm of diameter in the segment VII of the liver.
The tumor measured 18.5 x 16.0 x 12.5 cm, weighing 1869 grams, with a lobed surface of brown and white color. On the cuts, there was a solid brown appearance with cystic hemorrhagic areas; focal residues of cortical adrenal structures were noted at the periphery. The tumor presented small blue cells, multiple foci of tubular neuroepithelial differentiation, with primitive areas of nodular and diffuse arrangements and malignant features (Figure 2A). There were several foci of hyalinized and fibromyxoid stroma and areas of coagulative tumor necrosis, in addition to angiolymphatic infiltration. The neoplastic cells exhibited round or angular hyperchromatic nuclei and scarce clear cytoplasm; Flexner-Wintersteiner-type tubules and rosettes were observed, with central lumen formed by cells of same pattern (Figure 2 B). Immunohistochemistry study showed cytokeratin and CD99 (Figure 2 C and D), in addition to S-100 protein; whereas the samples submitted to the evaluation of 22q12 locus target sequences with a separation probe (LSI EWSR1 dual color) had a negative result (Figure 2 E). In addition to the negative result for rearrangement of the EWSR1 gene by the FISH method, the adrenal tumor also showed negativity for the NKX2.2 marker. Taken together, these characteristics contributed to rule out the initial hypothesis of PNET/ Ewing's sarcoma. Moreover, the tumor features were considered consistent with the diagnosis of immature malignant adrenal neoplasm of the type non-germ cell PNET. The treatment included 17 alternate cycles of chemotherapy with cyclophosphamide 600 mg/m2 [1080 mg IV (D1 and D2)] and vincristine 1.5 mg/m2 [2.7 mg IV (D1 and D2)], cycles: 2-4-6-8-10-12-14; cisplatin 20 mg/m2 [36 mg IV (D1 to D5)] and etoposide 100 mg/m2 [180 mg IV (D1 to D3)], cycles: 1-3-5-7-9-11-13. Two years after diagnosis, with BMI: 26.6 kg/m2, he had nonspecific symptoms and control evaluation was done. Abdominal image study (Figure 1D) showed a lesion with 15 cm of diameter in the segment VII of the liver, which was removed along with 70% of the organ.
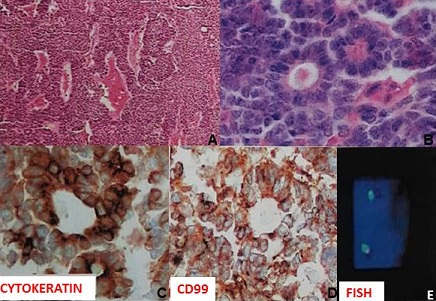
Figure 2: Histopathological features of the adrenal PNET.
(A) photomicrography of tumor sample showing small blue cells, multiple foci of
tubular neuroepithelial differentiation, primitive areas of nodular and diffuse
arrangements, and malignant features; (B) neoplastic cells with round or angular
hyperchromatic nuclei and scarce clear cytoplasm, in addition to
Flexner-Wintersteiner-type tubules and rosettes with central lumen formed by
cells of same pattern; (C) and (D): immunohistochemistry study showing
expression of cytokeratin and CD99; and (E): absence of rearrangement of the
EWSR1 gene by FISH method.
Histopathological findings revealed metastasis from the adrenal PNET of small blue cells. After hospital discharge, the patient had dyspnea and the chest X-ray showed pleural effusion. Three pleural drainages were then performed with removal of 1340 ml of a greenish colored fluid, due to a biliary fistula, which was successfully controlled by surgery. As the end time of the first chemotherapy cycles was longer than six months, the same protocol of the initial schedule was then reintroduced after he underwent the hepatectomy. Currently under ambulatory treatment, the patient maintains a good clinical condition.
Discussion
PNETs are rare types of cancer with origin in the neuroectoderm, which belong to the family of Ewing's sarcomas (ES) and are characterized by the presence of small round blue cells. Children and young adults are more often affected, and the outcome is poor in elderly [1-6]. The degree of neuronal differentiation has been used to sub classify these tumors, such as classic ES with minimal evidence of neuronal differentiation; and ES/PNET, which demonstrates this differentiation in several modalities of microscopic evaluation [4]. Translocation of t (11; 22) (q24; q12) involving the EWSR1 gene on chromosome 22 and the FLI-1 gene on chromosome 11 occur, yielding the EWL/FLI-1 fusion protein [1-7,
12]. Other change found in almost all PNETs is CD99, a very sensitive neural marker for this tumor, although not specific [4-6]. The gold standard diagnostic tool is the translocation showed by molecular techniques as FISH to detect the rearrangement of the EWSR1 gene [5]. Sometimes may be difficult to distinguish PNETs from other kidney cell-derived round cell tumors as the example of small cell carcinoma, small round cell desmoplastic tumor, or myxoid liposarcoma, because they can also express EWSR1 translocation [1-3,
5-8, 12]. In addition, neuroblastoma, malignant lymphoma, synovial sarcoma and Wilms tumor may be diagnostic challenges due to the morphological similarity of these tumors and the rarity of PNETs [5]. Adrenal PNETs should be also differentiated from more frequent conditions affecting this gland, including neuroblastoma, pheochromocytoma, and cancer of the adrenal cortex. Very important data for differential diagnosis of PNETs include the presence of small blue round cells, CD99 (+) membrane, and EWSR1 translocation [1-3,
5-8, 12]. The definitive diagnosis is hardly established prior to surgical intervention because it requires the routine combination of histology, immunohistochemistry and cytogenetic analysis [2]. PNETs can be found in the long and pelvic bones, as well as liver, kidneys, and adrenal glands [1,
2, 6-11]. In this patient the samples were submitted to immunohistochemical study, complemented with FISH evaluation. Initially, the possibility of classical ES/PNET was considered; however, some aspects were not consistent with this condition, as the negativity for NKX2.2 marker [12], and the absence of the EWSR1 gene rearrangement [1-3,
5-8, 12]. Therefore, the case study of an immature non-germ cell malignant adrenal PNET is herein described. It is noteworthy that based on the current literature this condition has been considered exceedingly uncommon.
Conclusions
Adrenal PNET is a very rare tumor of rapid growth and severe prognosis, with nonspecific manifestations. PNETs more often originates in primitive neuroectoderm of young people. Suspicion is based on histology, but must be confirmed by immunohistochemistry and cytogenetic analysis. Surgical resection and chemo irradiation is the best option for treatment.
Authors’ Contribution
VMS conceived the case study, participated in design,
coordinated the literature search and the analysis of data, interpreted the
results, prepared the draft manuscript, and coordinated the edition of the final
version
MCT participated in design, in the literature search, in the
analysis of data, and in edition of the final version
VVPS: participated in design, in the literature search, in
the analysis of data, and in edition of the final version
VMY: participated in design, in the literature search,
in the analysis of data, and in edition of the final version
MLD: participated in design, in the literature search, in the
analysis of data, and in edition of the final version
Conflicts of Interests
The authors declare that there are no conflicts of interests.
Ethical Considerations
Written informed consent was taken from the patient for publication of this case report.
Funding
None declared
References
1. Baldini EH, Demetri GD, Fletcher CD, Foran J, Marcus KC, Singer S. Adults with Ewing’s sarcoma/primitive neuroectodermal tumor adverse effect of older age and primary extraosseous disease on outcome: Ann Surg 1999; 230(1):79–86.
[Pubmed]
[PMC
Full text]
2. Carvajal R, Meyers P. Ewing's sarcoma and primitive neuroectodermal family of tumors. Hematol Oncol Clin North Am 2005; 19(3):501-25, vi-vii.
[Pubmed].
3. Delattre O, Zucman J, Melot T, Garau XS, Zucker JM,
Lenoir GM, Ambros PF, Sheer D, Turc-Carel C, Triche TJ, et al. The Ewing family of tumors - a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 1994; 331(5):294-9.
[Pubmed]
[Free
Full text].
4. Dong M, Liu J, Song Z, Li X, Shi T, Wang D,
Ren D, Chen J. Primary multiple pulmonary primitive neuroectodermal tumor. Case report and literature review. Medicine (Baltimore) 2015; 94(27):e1136.
[Pubmed]
[PMC
Full text].
5. Etani T, Naiki T, Ando R, Iida K, Naiki-Ito A, Takahashi S,
Kobayashi D, Kawai N, Tozawa K, Yasui T, Kohri K. A case of renal primitive
neuroectodermal tumor confirmed by fluorescence in situ hybridization. Case Rep
Oncol 2015; 8(1):205-11. [Pubmed]
[PMC
Full text]
6. Tsang YP, Lang BH, Tam SC, Wong KP. Primitive neuroectodermal adrenal gland tumour issues. Hong Kong Med J 2014; 20(5):444–6.
[Pubmed]
[Free
Full text].
7. Kumar S, Govinda V, Singh SK, Singh J. Bilateral adrenal PNET: a rare presentation. J Clin Diagn Res 2016;10(9):XD01-02.
[Pubmed]
[PMC
Full text].
8. Pal D, Chandra V, Ranjan KR, Chakrabortty D, Banerjee M. Ewing’s sarcoma of the adrenal. APSP J Case Rep 2016; 7(3):20.
[Pubmed]
[PMC
Full text].
9. Phukan C, Nirmal TJ, Kumar RM, Kekre NS. Peripheral primitive neuroectodermal tumor of the adrenal gland: a rare entity. Indian J Urol 2013; 29(4):357-9.
[Pubmed]
[PMC
Full text].
10. Yoon JH, Kim H, Lee JW, Kang HJ, Park HJ, Park KD, et al. Ewing sarcoma/peripheral primitive neuroectodermal tumor in the adrenal gland of an adolescent: A Case report and review of the literature. J Pediatr Hematol Oncol. 2014; 36(7):e456-9.
[Pubmed].
11. Zhang L, Yao M, Hisaoka M, Sasano H, Gao H. Primary Ewing sarcoma/primitive neuroectodermal tumor in the adrenal gland. APMIS 2016; 124(7):624-9.
[Pubmed].
12. Sarver AE, Sarver AL, Thayanithy V, Subramanian S. Identification, by systematic RNA sequencing, of novel candidate biomarkers and therapeutic targets in human soft tissue tumors. Lab Invest 2015; 95(9):1077-88.
[Pubmed]
[Free
Full Text].

