Case Report

Ectopic ACTH Secretion by Islet Cell Neuroendocrine Carcinoma: Case Report and Review of the Literature
Marcos M. Chertman, Lila S. Chertman
University of Miami Hospital, University of Miami Miller School of Medicine, Mt. Sinai Medical Center Miami Beach, Miami, USA
Submitted June 19, 2013;Accepted July 26, 2013, Published: July 26, 2013
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Objective
To review the literature and describe a case of ectopic Cushing’s syndrome caused by a neuroendocrine carcinoma of the pancreas.
Methods
We present the clinical, laboratory, radiologic, and pathologic details of the case and review the relevant literature.
Results
A 65 year old woman presented with Cushing’s syndrome, hyperglycemia, hypokalemia, and a pancreatic mass. CT-guided aspiration and laparoscopic distal pancreatectomy were performed. Pathological examination revealed a neuroendocrine carcinoma with ectopic production of ACTH. She was treated initially with Ketoconazole and then monthly Sandostatin, and Cushingoid features have resolved.
Conclusion
Pancreatic neuroendocrine tumors that secrete ACTH causing Cushing’s syndrome are rare, posing a diagnostic and therapeutic challenge to the practitioner. We present a case of a rapidly evolving neuroendocrine Cushing’s syndrome that was treated successfully with surgery and medical management.
Key Words
Pancreas, Neuroendocrine, Ectopic, Cushing’s syndrome, Octreotide
Introduction
Pancreatic neuroendocrine tumors (pNETs) are neoplasms expressing Chromogranin A and synaptophysin [1, 2, 3]. PNET’s are rare, with an incidence of 1/100,000 per year, making up 1-3% of all Pancreatic tumors, and accounting for approximately 1000 new cases in the USA every year [1, 3, 4].They arise from multipotential stem cells in the ductal epithelium and reports suggest an increased incidence and prevalence in the last several years [4, 5]. Functioning pancreatic NETs cause clinical syndromes secondary to hypersecretion of hormones and are defined on the basis of clinical symptoms rather than immunohistochemical findings. Most have already metastasized to the liver by the time of diagnosis [4, 5, 6]. Ectopic ACTH secretion (EAS) leading to Cushing’s syndrome is rare and is responsible for 4%-18% of ACTH-dependent cases [6]. When the syndrome was first reported the most common etiology was small-cell lung cancer [3]. Over the next 4 decades multiple other tumors have been described including bronchial, pancreatic, and medullary [4, 5, 6]. EAS may be associated with rapidly evolving highly malignant tumors, or with other less aggressive NETs, exhibiting variable features from the typical findings of CS to infectious, reproductive, orthopedic, and even psychiatric findings [7]. EAS secondary to an ACTH-producing pNETs is a particularly aggressive disorder with early metastases even before the development of Cushing’s syndrome and accounts for 10% of cases of EAS [7]. ACTH released by a pancreatic tumor enters the enterohepatic circulation and is rapidly metabolized by the liver, whereas ACTH released by bronchial, thymic, and ovarian neuroendocrine tumors directly enter the systemic circulation and result in Cushing’s syndrome. Therefore, patients with an ACTH-secreting pancreatic neuroendocrine tumor causing Cushing’s syndrome almost always already have liver metastases [12,13]. We present the case of a woman with rapidly evolving Cushing’s due to a neuroendocrine carcinoma of the pancreas.
Case Presentation
A 65-year-old woman with a 4 year history of previously controlled hypertension was admitted twice to an outside hospital with dizziness, muscle weakness, unexplained hypokalemia, and paroxysmal elevation of blood pressure requiring multiple adjustments of her medications. Other complaints included facial swelling and weight gain, but no diagnosis was established. It was only after the patient’s discharge that her daughter, a nurse, demanded endocrine evaluation because she had noticed increased facial hair. Laboratory findings from that evaluation included cortisol 21.3, ACTH 150 mcg, free-T4 0.84, FSH 0.9, LH < 0.10, 24 hour urinary free cortisol 1090 mg, and lack of suppression of cortisol after high dose Dexamethasone. MRI of the brain was unremarkable. She was admitted to our service 4 months later with generalized weakness, hyperglycemia (glucose 700), and hypokalemia (2.1). On physical exam, blood pressure was 170/110, pulse 90 and regular. Cushingoid features included: facial plethora, hirsutism, abdominal striae and ecchymoses, atrophic skin, truncal obesity, and 1+ edema of the ankles. Treatment was started with fluids, IV insulin, and potassium replacement (Table 1, figure 1). CT abdomen and pelvis showed a low density lesion of the liver measuring 1.1 x 1.3 cm and a 1.2 x 1.2 cm calcified mass within the pancreatic tail. Octreotide scan showed questionable area of uptake in the mid central liver and the right hepatic lobe. CT-guided aspiration of the mass in the pancreas showed neuroendocrine-type cells positive for chromogranin A and synaptophysin.
Table 1: Preoperative findings
|
Salivary Cortisol
|
24 hours UFC
|
Metanephrine
|
Normetanephrine
|
Chromogranin A
|
ACTH
|
High dose DEXA 8 mg
|
5-HIAA
|
|
1.427 mcg/dL
|
2257 mcg/24 hours
|
13
|
21
|
9
|
228.4 pg
|
Not suppressed
|
7.7
|
UFC: Urinary free cortisol; ACTH: Adrenocorticotropic hormone; DEXA
dexamethasone; 5-HIAA: hydroxyindoleacetic acid
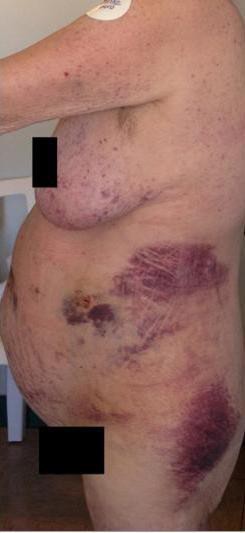
Figure 1: Clinical Presentation
Surgical findings
Laparoscopic distal pancreatectomy and pathology results were consistent with a 1.5 cm neuroendocrine carcinoma (mixed small and large cell) with ectopic production of ACTH and synaptophysin with metastasis to 1 of 2 lymph nodes and the liver. (Table 2, figures 2 & 3)
Table 2: Postoperative findings
|
Postoperative day
|
AM cortisol
|
PM cortisol
|
24 hours urinary cortisol (mcg/24 hours)
|
ACTH
|
|
4th day
|
59.42
|
64.71
|
-
|
280.7
|
|
8th day
|
24.36
|
38.86
|
510
|
-
|
|
1 month
|
10.75
|
10.85
|
50
|
118.6
|
Because cortisol and ACTH levels remained elevated she was sent home on ketoconazole. Four weeks later she no longer required insulin and blood pressure was controlled without medications. Potassium levels were within normal limits on 60 meq KCL. She later presented with cough, fever, and dyspnea leading to admission to another institution for pneumonia. She was discharged with antibiotics but developed nausea, diffuse abdominal pain, and diarrhea and was admitted to our institution where blood cultures were negative and chest x-ray showed interstitial markings with coarse ill-defined liner 4densities and ground-glass opacities. Antibiotic therapy and ketoconazole were withheld and improvement was noted over the next 48 hours.
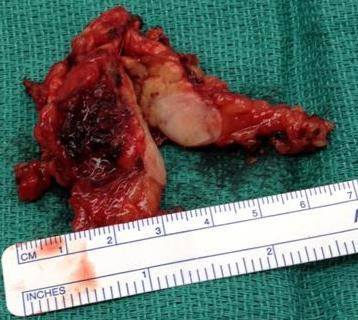
Figure 2: Resected specimen
As an outpatient, monthly Sandostatin LAR 30mg was started because of elevated ACTH and cortisol.
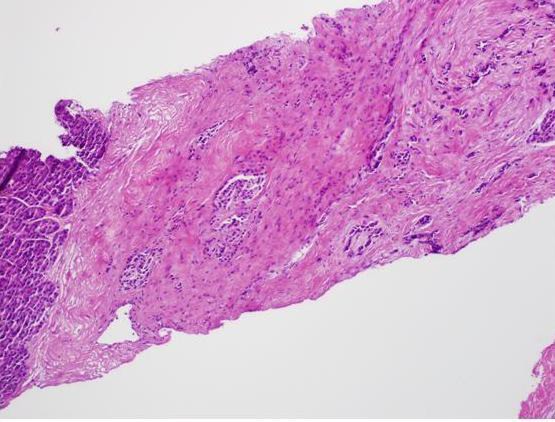
Figure 3: Photomicrograph
Improvement was noted with almost complete resolution of Cushingoid features. Her most recent MRI of the liver is normal and we have kept her on Sandostatin LAR with close follow up of hormonal levels and liver imaging.
Discussion
In Clark’s 1984 review of the known 42 cases of pancreatic tumors associated with Cushing’s syndrome, he found that metastases occurred in 88% of the patients and 60% were dead in 2 years or less [8]. A 1994 case series at the National Cancer Institute by Doppman et al, of patients with Cushing syndrome secondary to ACTH production by a pancreatic tumor demonstrated liver metastases at presentation in each of the 10 cases [7]. According to the National Cancer Institute Surveillance, Epidemiology, and End Results (SEER) Program database, a total of 35,825 NETs were reported over a 30 year period (1973 – 2004) in the United States with a significant increase in the annual age-adjusted incidence of NETs [5]. This increase may in part be due to improvements in classification of the NETs and advances in accessibility of diagnostic imaging [5] .
Pathology
Most pancreatic NETs are well-demarcated, with < 20 mitoses per high power field and can be identifiedusing antibodies against chromogranin A and synaptophysin, CEA, and CA 19-9 [9].
Immunohistochemical labeling for Ki67 with MlB1 antibody can be used to determine their proliferative rate. Pancreatic NECs, which average a diameter of 4 cm, have ill-defined borders with hemorrhage and necrosis. By definition they there are > 20 mitoses per 10 HPF, and some express p53. They are classified as small cell NEC or large cell NEC depending upon the size of the neoplastic cells, prominence of nucleoli, and amount of cytoplasm. Large cell NECs label for chromogranin A and synaptophysin, but small cell NECs may not [9] Pancreatic NETs can be part of genetic syndromes like multiple endocrine neoplasia type 1 (MEN1), von Hippel Lindau (VHL), tuberous sclerosis, and neurofibromatosis, although most pNETs are sporadic [4, 9]. About 80% of patients with MEN1 have pancreatic NETs like gastrinomas or insulinomas, which occur at an earlier age than do sporadic pNETs. Management of pNETs associated with these genetic syndromes may be different than that of sporadic pNETs since these are predisposed to multifocal pancreatic and extra-pancreatic neoplasms [10]. Stratifying patients into prognostic groups has been complicated by the use of several different classification systems that are independently validated and prognostic for survival, including the American Joint Committee on Cancer/International Union against Cancer (AJCC) and the European neuroendocrine Tumor Society (ENETS) [9, 11]. ENETS classifies pancreatic NETs into a four-stage TNM 5 classification, and a three-tier grading classification (low G1, intermediate G2, and high grade G3) based on mitoses and the Ki67 labeling rate. The AJCC TNM staging distinguishes between localized tumors (stage 1), locally advanced resectable tumors (stage II), locally advanced unresectable tumors (stage III), and distantly metastasized tumors (stage IV) but does not include tumor grading guidelines [9] (Tables 3 & 4).
Table 3: WHO classification of pancreatic endocrine tumors [4]
|
Tumor
|
Size (cm)
|
Mitotic count (per 10 HPF)
|
Ki-67 index
|
Angioinvasion
|
Metastasis
|
|
Well differentiated endocrine tumor
|
|
|
|
|
|
|
Benign
|
<2
|
<2
|
<2%
|
Absent
|
Absent
|
|
Uncertain
|
>2
|
2-10
|
>2%
|
Present
|
Absent
|
|
Well differentiated endocrine carcinoma
|
>2
|
<10
|
>2%
|
Present
|
Present
|
|
Poorly differentiated endocrine carcinoma
|
|
>10
|
>20%
|
Present
|
Present
|
In 2010 the World Health Organization introduced a grading classification based on morphological criteria and the assessment of proliferation fraction using the Ki67 index [12]. They defined a NET as a well-differentiated neuroendocrine neoplasm expressing neuroendocrine markers (chromogranin A and synaptophysin) with mild to moderate nuclear atypia, < 20 mitosis per 10 HPF, with grade G1 to G2. A NEC is a poorly differentiated high grade malignant neoplasm that is always grade G3, with small to large cells expressing neuroendocrine differentiation, and marked nuclear atypia, >20 mitoses per 10 HPF [12, 13].
Table 4: TNM system of staging for pancreatic neuroendocrine tumors [4]
|
Primary Tumor
|
Size (cm)
|
|
Tx
|
Primary tumor can
not be assessed
|
|
T1
|
<2 cm limited to
pancreas
|
|
T2
|
2-4 cm, limited to
pancreas
|
|
T3
|
Beyond pancreas, no
invasion of celiac axis or superior mesenteric artery
|
|
T4
|
Beyond pancreas,
invasion of celiac axis or superior mesenteric artery
|
|
Regional lymph nodes
|
|
|
Nx
|
Regional lymph nodes
can not be assessed
|
|
N0
|
No regional lymph
node metastasis
|
|
N1
|
Regional lymph node
metastasis
|
|
Distant metastasis
|
|
|
Mx
|
Distant metastasis
cannot be assessed
|
|
M0
|
No distant
metastasis
|
|
M1
|
distant metastasis
|
|
Staging
|
|
|
Stage Ia
|
T1N0
|
|
Stage Ib
|
T2N0
|
|
Stage IIa
|
T3N0
|
|
Stage III
|
T4NanyM0
|
|
Stage IV
|
TanyNanyM1
|
Other than neuroendocrine microadenomas, all pancreatic NETs are considered to have malignant potential [6, 9]. Excluding insulinomas, the 5-year survival for patients with pNETs is 65% and the 10- year survival is 45%. Stage (extent of disease) and grade (based on proliferative rate) are most helpful in stratifying pNETs into different risk groups. Pancreatic NECs, as opposed to pancreatic NETs, display highly aggressive behavior and are usually unresectable at diagnosis with invasion of nearby tissues, widespread metastases, and survival ranging from 1 month to 1 year [9].
Imaging
The initial imaging study used to identify and stage a pNET is a CT with contrast, which displays the tumor as a hyperdense mass [4]. MRI, which has the added benefit of not requiring IV contrast, is especially useful for smaller tumors which display high signal intensity on the T2 weighted images, versus low signal intensity on T1 weighted images [4, 14].
Octreoscan and PET are functional imaging techniques especially useful for patients with extrahepatic tumor spread and help guide treatment options. Because pNETs express many somatostatin receptors on their cell surfaces they uptake the somatostatin tracer, or its longer lasting analog octapeptide, allowing for tumor identification [4, 10, 13, 14]. It is indicated as the first staging procedure and as screening for extrahepatic disease [13]. However, false positives occur, nonfunctional tumors are less frequently visualized, and tumors < 1cm are missed in >50% of cases [6, 14].
Treatment
The only potential for cure of pNETs is complete surgical resection [4]. In patients with metastases, debulking of tumor mass is valuable in combination with multimodality treatment [15]. Surgical debulking is appropriate if at least 90% of the visible tumor can be removed, which may reduce hormonal symptoms [4]. Approaches to liver metastases include debulking, embolization, and ablation. Cytoreductive techniques, reserved for unresectable liver metastases smaller than 5 cm, include radiofrequency ablation, cryoablation, microwave ablation, and alcohol ablation [3, 4]. Hepatic artery embolization, based on the principle that liver tumors derive their blood supply from the hepatic artery 6 rather than the portal vein like normal hepatocytes do, can be used as palliative therapy either alone or with intra-arterial infusion of doxorubicin, cisplatin, or a radionuclide microsphere [13]. Liver transplantation for patients with unresectable liver metastases is rare due to early disease recurrence and high postoperative mortality [4].
Octreotide and Lanreotide are longer acting analogues of human somatostatin, which inhibit endocrine activity [3]. Octreotide has been effective in controlling hormonal secretion and prolonging time to progression in 50% of patients with metastatic NETs [4, 16]. NETs overexpress somatostatin receptor subtypes, confirmed by OctreoScan showing that 80% of pNETs express somatostatin receptors on their surface [4]. Octreotide has anti-proliferative effects that stabilize tumor growth via suppression of VEGF, IGF, and growth hormone [3]. In patients with G1 and G2 tumors, Octreotide is the first line of treatment, especially when there are hormonal related symptoms [3, 6]. Octreotide’s action on hormonal secretion is mainly through SSTR-2, while tumor growth suppression and apoptosis action occurs through SSTR-3 and 5 [4, 6]. Pasireotide, the newly released somatostatin analogue, targets 4 of the 5 somatostatin receptors, with the highest affinity for subtype-5 [3, 6].
The somatostatin-receptor complex partially internalizes after binding occurs, providing a means to deliver cytotoxic treatments with agents like indium (111In), yttrium (90Y), and lutetium (177Lu), in a new treatment approach known as targeted radiopeptide therapy (TRT) or peptide receptor radionuclide therapy (PRRT) [17]. Its unique features include highly specific binding of the radio labeled hormone analog to its target, fast clearance of residual radioactivity, and long retention of radioactivity in the tumor cells . TRT/PRRT used to treat SSTR-2 positive tumors appears to be the most effective therapeutic option in unresectable NETS where local and regional treatment options are no longer feasible, although the best peptide, radionuclide, and time frame for starting treatment has yet to be determined [17]. Interferon-alpha has also been used both alone and in conjunction with octreotide to palliate hormonal symptoms and has been shown to have antitumor effects by inhibiting angiogenesis, stimulating T cells, and inducing cell cycle arrest but its toxic side effects make it unpopular [3]. Systemic treatments include streptozotocin but it is rarely used secondary to its toxicity [4]. Everolimus, an oral inhibitor of the mammalian target of rapamycin (mTOR), has recently been approved for the treatment of unresectable pancreatic neuroendocrine carcinomas. Because pNETs are highly vascular, angiogenesis inhibitors like Suantinib and Bevacizumab are being investigated, and Sunatinib has been recently approved for treatment of metastatic pNETs [3]. For patients with Cushing’s syndrome as a result of pNETs, adrenal-blocking agents like ketoconazole and metyrapone are used prior to adrenalectomy [6]. Octreotide may be effective in controlling the hormone excess state, and may be used alone or in combination with alpha-interferon. In our patient, cortisol and ACTH levels remained elevated postoperatively, and we started Ketoconazole, an antifungal with steroid blocking action, which was effective in controlling excess cortisol secretion but due to side effects was discontinued. Sandostatin LAR 30 mg monthly was started which has been effective [6].
Diagnostic investigation is initiated by symptomatology, but in the vast majority of patients this is already indicative of metastasis and the delay in diagnosis of NETs from first symptoms is an average of 7 years [18]. In Strosberg et al’s series of 123 patients, they recommend radiologic surveillance continue past 5 years to detect late recurrences which occur in 35% of patients, although the peak recurrences occurred 2 years postoperatively [11]. The ENETS Consensus guidelines for management of pNETs published in 2012 recommends follow-up at 3 to 6 month intervals in those with metastatic disease, or yearly in those with localized disease, with hormone levels, specific markers, and contrast enhanced CT or MRI and SRS [6].
Conclusions
We have reported a fascinating case of a patient with rapidly evolving Cushing’s syndrome, caused by pNEC of the pancreas with liver metastasis, whose clinical course has responded to excision of the pancreatic tumor, use of ketoconazole and most recently Octreotide-LAR therapy.
Learning points
It is important to consider Cushing’s syndrome in the differential diagnosis of unexplained hypokalemia and hypertension. A thorough history and physical, including interviewing the patient’s family members, yield vital clues to the diagnoses. Early recognition of Cushing’s syndrome and its etiology may allow for better treatment outcome and prevent complications. Neuroendocrine tumors of the pancreas, although recently increasing in incidence, are a rare cause of Cushing’s syndrome and the presence of liver metastases at the time of diagnosis is common.
Ethical Considerations
Consent for publication was obtained from the patient by Dr. Marcos Chertman and is provided in a separate file.
Competing Interests
The authors have no multiplicity of interest to disclose.
Author’s contributions
MCMC carried out the literature search, prepared the case report and conclusions draft, and edited the final manuscript. LC prepared the literature review and discussion draft, and edited the final manuscript.
Both authors read and approved the final manuscript for submission.
Acknowledgements
The authors wish to thank Dr. Prodipto, M.D. PhD, PGY3 in pathology at Jackson Memorial Hospital, for the pathology slides. We also would like to thank Dr. Kuker M.D. of the radiology department at the University of Miami Health System for the radiology images. We thank Dr. Reginald Pereira for kindly referring the patient.
Abbreviations
CS, Cushing’s syndrome; CT, computed tomography; EAS, ectopic secretion ofACTH, pNET, pancreatic neuroendocrine tumor; pNEC, pancreatic neuroendocrine carcinoma; ACTH, adrenocorticotropic hormone; 5-HIAA, 5 hydroxyindoleacetic acid; UFC, urinary free cortisol; TIA,transient ischemic attack; MRI, magnetic resonance imaging; CgA, chromogranin A; MEN1, multipleneuroendocrine neoplasia type 1; VHL, von hippel landau; HPF, high power field; mTOR,mammaliantarget of rapamycin.
References
[1]Strosberg JR, Cheema A, Weber J, et al. Prognostic validity of a novel American Joint Committee on cancer staging classification for pancreatic neuroendocrine tumors. Journal of Clinical Oncology 2011; 29: 3044-3049.[pubmed]
[2].Strosberg JR, Nasir A, Hodul P, Kvols L. Biology and treatment of metastatic gastrointestinal neuroendocrine tumors. Gastroinstest Cancer Res 2008; 2(3):113-125.[pubmed]
[3]Strosberg JR, Cheema A, Kvols L. A review of systemic and liver-directed therapies for metastatic neuroendocrine tumors of the gastroenteropancreatic tract. Cancer Control 2011; 18 (2): 127-137.[pubmed]
[4].Milan SA and Yeo CJ. Neuroendocrine tumors of the pancreas. Curr Opin Oncol 2012; 24: 46-55.[pubmed]
[5].Yao JC, Hassan M, Phan A, et al. One hundred years after “Carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol 2008; 26: 3063-3072.[pubmed]
[6].Jensen RT, Cadiot G, Brandi ML, et al. ENETS consensus guidelines for the management of patients with digestive neuroendocrine neoplasms: functional pancreatic endocrine tumor syndromes. Neuroendocrinology 2012; 95: 98-119.[pubmed]
[7].Doppman JL, Nieman LK, Cutler GB, et al. Adrenocorticotropic hormone-secreting islet cells tumors: are they always malignant? Radiology 1994; 190: 59-64.[pubmed]
[8].Clark ES, Carney JA. Pancreatic islet cell tumor associated with Cushing’s syndrome. Am J. Surg Pathol 1984; 8: 17-24.[pubmed]
[9].Klimstra DS, Arnold R, Capella C, et al. Neuroendocrine tumours of the pancreas. In: WHO classification of tumours of the digestive system, 4th ed, Bosman TF, Carneiro F, Hruban RH et al (Eds), International Agency for Research on Cancer ( IARC), Lyon 2011. p. 44-48.
[10].Ellison TA, and Edil BH. The current management of pancreatic neuroendocrine tumors. Advances in Surgery 2012; 46: 283 – 296. [pubmed]
[11].Strosberg JR, Cheema A, Weber JM, et al. Relapse-free survival in patients with nonmetastatic, surgically resected pancreatic neuroendocrine tumors: an analysis of the AJCC and ENETS staging classifications. Annals of Surgery 2012; 256: 321-325. [pubmed]
[12].Rindi G, Arnold R, Bosman FT, et al. Nomenclature and classification of neuroendocrine neoplasms of the digestive system. In: WHO Classification of Tumours of the Digestive System, 4th ed, Bosman TF, Carneiro F, Hruban RH, Theise ND (Eds), International Agency for Research on cancer (IARC), Lyon 2010. p.13.
[13].Falconi M, Bartsch DK, Eriksson B, et al. ENETS consensus guidelines for the management of patients with digestive neuroendocrine neoplasms of the digestive system: well-differentiated pancreatic non- functioning tumors. Neuroendocrinology 2012; 95: 120-134.[pubmed]
[14].Bushnell DL and Baum RP. Standard imaging techniques for neuroendocrine tumors. Endocrinol Metab Clin N Am 2010; 40: 153-162.[pubmed]
[15].Sarmiento JM, Heywood G, Rubin J, et al. Surgical treatment of neuroendocrine metastases to the liver: A plea for resection to increase survival. J Am Coll Surg 2003; 197: 29-37. [pubmed]
[16].Nicolas G, Giovacchini G, Muller-Brand J, and Forrer F. Targeted radiotherapy with radiolabeled somatostatin analogs. Endocrinol Metab Clin N Am 2011; 40: 187 – 204. [pubmed]
[17].Kwekkeboom DJ, de Herder WW, Krenning EP. Somatostatin receptor-targeted radionuclide therapy in patients with gastroenteropancreatic neuroendocrine tumors. Endocrinol Metab Clin N Am 2011; 40:173 -185. [pubmed]
[18].Modlin IM, Champaneria MS, Chan AK, Kidd M. A three-decade analysis of 3,911 small intestinal neuroendocrine tumors: the rapid pace of no progress. Am J Gastroenterol 2007; 102: 1464 - 1473.[pubmed]

