Case Report

Role of Computed Tomography Imaging in Evaluation of Neurofibromatosis Type -2
* Sharad Pandey, * Kulwant Singh, * Vivek Sharma,* Richa Singh Chauhan
- *Neurosurgery, Institute of Medical Sciences, Banaras Hindu University, Uttar Pradesh, Varanasi, India
- Submitted: Sunday, August 9, 2015
- Accepted: Friday, December 18, 2015
- Published: Tuesday, January 26, 2016
This is an Open Access article distributed under the terms of the Creative Commons Attribution License ((http://creativecommons.org/licenses/by/3.0)which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited
Abstract
Neurofibromatosis type 2 (NF-2) is a rare, inherited, tumor-prone disorder, characterized by multiple neoplasms of the central and peripheral nervous system associated with ocular abnormalities. It is associated with tumors of Schwann cells and meninges. Affected individuals inevitably develop schwannomas, usually affecting both vestibulocochlear nerves. In this article, we present two cases of sporadic NF2 with computed tomographic findings of bilateral acoustic schwannomas and multiple intracranial meningiomas with right orbital cavernous angioma.
Key words
Neurofibromatosis type 2, Bilateral Acoustic schwannoma, Meningioma, Cavernous Angioma, Computed Tomography
Introduction
Neurofibromatosis (NF) is an inherited autosomal dominant disease mainly affecting the skin, nervous and musculoskeletal systems, divided into two types: NF-1 & NF-2.Neurofibromatosis type 2 (NF-2) is a rare (as compared to NF-1) genetic disorder with prevalence of about 1 in 60,000 [1] and has almost equal sex distribution. It is characterized by the development of schwannomas and multiple meningiomas. This disorder can be diagnosed when a pathogenic mutation in the NF2 gene (on long arm of chromosome 22) is identified or when the diagnostic criteria proposed by Manchester are fulfilled (Table 1) [2]. Compared to NF-1, patients with NF-2 may have cutaneous schwannomas, but they rarely have café -au-lait spots or cutaneous neurofibromas [2]. NF2 might be difficult for the clinicians to diagnose because cutaneous manifestations are often less or absent. Therefore the diagnosis is usually made by the radiologists on imaging.
Table 1:
Diagnostic Criteria proposed by
Manchester[2]
|
A patient who meets
condition A, condition B, condition C, or condition D has NF2.
A. Bilateral VS
B. 1st degree family
relative with NF2
and
unilateral VS or any two
of neurofibroma, meningioma,
glioma, schwannoma, or
posterior subcapsular lenticularopacities
C. Unilateral V
and
any two of neurofibroma,
meningioma, glioma, schwannoma,
or posterior subcapsular
lenticular opacities
D. Multiple meningiomas
(two or more)
and
unilateral VS or any two
of neurofibroma, glioma,schwannoma, or cataract
|
Case 1:
A 19 years old female patient presented in the Neurosurgery OPD with complaints of headache since three months generalized tonic clonic seizures since 2 months with unilateral hearing loss &tinnitus (left ear). There was no history of fever, otalgia, ear discharge or trauma. No family history of this neurocutaneous disorder was present. On physical examination she was well oriented with no evidence of any skin lesion. Computed tomography (CT) scan of Brain showed multiple relatively defined, extraaxial, hyperdense mass lesion in right parietal parasagittal region and left occipital region (en-plque type). Largest one of them measuring 2.2 cm x 2.2 cm (Anteroposterior x Transverse). Few of them showed internal calcification foci. On contrast study, the lesion showed homogenous enhancement suggestive of
meningiomas. The meningeoma in left occipital region caused overlying bony hyperostosis. Relatively defined, isodense, heterogeneous, extraaxial mass lesions were noted at bilateral CP angles (Left larger then right, measuring approximately 3.2 cm x 3.4 cm) extending into respective internal acoustic canals causing canal widening. On contrast study, the lesion showed mild heterogeneous enhancement suggestive of Acoustic schwannomas (Figure 1).
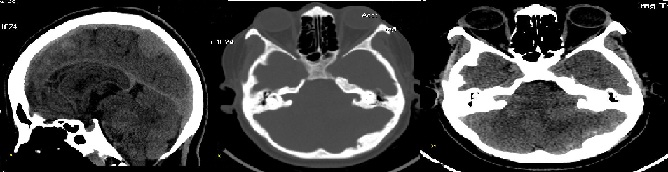
Figure 1:(a) CECT head showing multiple extraaxial moderately enhancing lesion with few calcification (b) bony hyperostosis in left occipital region (c) extraaxial mild enhancing lesion at bilateral CP angle.
Case 2:
18 years old female patient presented in the Neurosurgery OPD history of progressive bilateral hearing loss, which was unilateral at onset along with generalized tonic clonic seizures for 2 years and gradually increasing right eye proptosis with diplopia. There was no history of fever, otalgia, ear discharge or trauma. No family history of this neurocutaneous disorder was present. On physical examination two skin tags on right upper arm were noticed, with no other significant abnormality (not evaluated further). Cranial Computed tomography (CT) scan showed multiple relatively defined, extraaxial, hyperdense mass lesions in right occipital , left frontal and left temporal lobes and in interhemispheric space between bilateral frontal lobes, largest measuring 2.8 cm x 2.6 cm and few showed internal calcification foci. On contrast study, the lesion showed mild to moderate enhancement suggestive of Meningiomas. Relatively defined, hyperdense, extraaxial mass lesion were noted at bilateral CP angles extending into respective internal acoustic canals causing canal widening. On contrast study the lesion showed moderate enhancement suggestive of Acoustic schwannomas. An ill-defined hyperdenseextraaxial mass lesion was noted that appears to be attached to right medial orbital wall showing a focus of internal calcification and no evidence of ipsilateral optic nerve involvement. On contrast study, the lesion showed moderate enhancement suggestive of right orbital cavernous angioma (Figure 2).
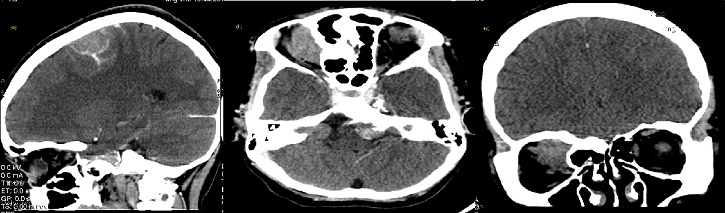
Figure 2: (a) CECT head showing multiple extraaxial mild to moderate enhancing lesion with few calcification (b) extraaxial moderately enhancing lesion at bilateral CP angle (c) extraaxial moderately enhancing lesion attached to right medial orbital wall.
Discussion
Neurofibromatosis 2 is a rare autosomal dominant neurocutaneous disorder. The term MISME has been proposed to the NF 2 syndrome, due to multiple inherited schwannomas (MIS), meningiomas (M) and ependymomas (E) [3]. Bilateral vestibulocochlear nerve schwannomas are considered as the hallmark of NF-2 even without biopsy confirmation. This usually presents in the second or third decade of life, with a peak in the 20s. It has varied clinical manifestations but approximately 30-45% of patients are diagnosed because of symptoms resulting from vestibulocochlear nerve (CN VIII) schwannomas, such as hearing loss, tinnitus, imbalance and weakness due to facial nerve (CN VII) involvement. Often, the first clinical sign of NF-2 is sudden hearing loss due to the development of bilateral or unilateral vestibular schwannomas. Reason being the vestibular schwannomas is symptomatic at a relatively small size and cause symptoms by compressing or stretching the cochlear nerve, affecting the vascular supply to the nerve or to the cochlea, or causing hemorrhage into the nerve or cochlea [4 5].In addition to CN VIII, schwannomas of other cranial nerves may also occur. Presence of Oculomotor, Trochlear or Abducent nerve tumor should raise the suspicion of NF-2. Similarly, involvement of more than one cranial nerve warrants work up for NF-2. Multiple intracranial (including optic nerve) and intraspinal meningiomas, are also common in this disorder. Other associated intracranial lesions are non-neoplastic calcifications, most common being the choroid plexus calcification. However, cerebellar and cerebral cortical calcifications may also be present [6].
Some low-grade central nervous system malignancies (ependymomas) may also develop. Other than cranial tumors, ophthalmic features are also prominent which include reduced visual acuity and cataract (posterior subcapsular lenticular opacity/ juvenile cortical cataract). About 70% of NF-2 patients have skin tumors (intracutaneous plaque-like lesions or more deep-seated subcutaneous nodular tumors).
Management of NF- 2
Treatment of manifestations: Treatment of vestibular schwannoma is primarily surgical; stereotactic radiosurgery, alternative to surgery may be with the gamma knife. Persons with vestibular tumors should be aware of insidious problems with balance and underwater disorientation, which can result in life threatening situation[2, 7].Treatment for hearing loss includes referral to an audiologist, lip-reading and sign language instruction, and possibly hearing aids and/or cochlear or brainstem implants.
Surveillance: For affected or at-risk individuals, annual magnetic resonance imaging (MRI) beginning at the age of approximately 10 to 12 years and continuing until at least the fourth decade of life and hearing evaluation, including auditory brainstem evoked response testing [8, 9].
Agents/circumstances to avoid: Radiation therapy of NF2-associated tumors, especially in childhood, may induce, accelerate, or transform tumors.
Testing of relatives at risk: Early identification of relatives who have inherited the family-specific NF2 mutation allows for appropriate surveillance, resulting in earlier detection and treatment of disease manifestations.
Our patients had bilateral CN VIII schwannomas with intracanalicular extension, multiple intracranial meningiomas and onerightintraorbital (intraconal) tumor (cavernous hemangioma on imaging). The schwannomas were the cause of their hearing loss while multiple meningiomas were responsible for headache and seizures. Although they had no family history, CT finding of bilateral schwannomas is considered diagnostic for this syndrome, even without the need of a biopsy. However, spinal MRI screening did not reveal any tumor in both the cases. At least two thirds of individuals with NF 2 develop spinal tumors, which are often most devastating and difficult to manage [10, 11, 12]. Spinal lesions in NF 2 include Schwannomas which are more common [10] and Meningiomas. Based on these findings, the diagnosis of NF 2 was considered.
Conclusion
NF2 represents a difficult management problem with most patients facing substantial morbidity and reduced life expectancy. Although, watchful waiting with careful surveillance have a role, but in symptomatic patients surgery remains the focus of current management with CT imaging being an important diagnostic tool.
Abbreviations
NF2: Neurofibromatosis 2
MRI: Magnetic resonance imaging
CNS: Central nervous system
CN: Cranial nerve
Computed tomography
OPD: Out patient department
Conflict of Interests
The authors declare that there are no conflict of interests.
Authors Contribution
SP: Concept and design, drafting of the article.
KS: Critical revision of the article for important intellectual content.
VS : Editing and final approval of the article.
RSC: Collection and assembly of data and preparation of manuscript.
Ethical Consideration
Written informed consent was obtained from both the patients for publication of their case. A copy of consent is available with the authros.
Funding
None declared
References
[1].Evans DG. Neurofibromatosis type 2 (NF2): A clinical and molecular review. Orphanet J Rare Dis 2009;4:16.[Pubmed]
[2].Evans DGR, Huson S, Donnai D, Neary W, Blair V, Newton V, Harris R: A clinical study of type 2 neurofibromatosis. 1992; 84:603-18.
[3].Gangadhar K, Kumar S, Bhatia L, Agarwal A. A Complete Constellation of Nervous System Lesions of NF2: Imaging Evaluation. Case Reports in Radiology. 2012; 2012:353179. doi:10.1155/2012/353179.
[4].Baser ME, Evans DG, Gutmann DH. Neurofibromatosis 2. CurrOpin Neurol. 2003; 16:27-33.[Pubmed]
[5].Spilberg G, Marchiori E, Gasparetto EL, Cabral RF, Takayassu TC, Batista RR et al. Magnetic resonance findings of neurofibromatosis type 2: a case report. Cases J. 2009; 2:6720-7.[Pubmed]
[6].Osborn Anne G. Disorders of Histogenesis: Neurocutaneous Syndromes. In: Diagnostic Neuroradiology. Mosby Year Book, 1994: 84 - 93.
[7].Kanter WR, Eldridge R, Fabricant R, Allen JC, Koerber T. Central neurofibromatosis with bilateral acoustic neuroma: genetic, clinical and biochemical distinctions from peripheral neurofibromatosis. Neurology1980; 30:851–859.
[8].Evans DGR, Newton V, Neary W, etal.Use of MRI and audiological tests in pre-symptomatic diagnosis of type 2 neurofibromatosis (NF2). J Med Genet2000; 37:944–947.[Pubmed]
[9].Evans DGR, Baser ME, O'Reilly B, et al.Management of the patient and family with Neurofibromatosis 2: a consensus conference statement. Br J Neurosurg 2005; 19:5–12.[Pubmed]
[10].Mautner VF, Tatagiba M, Lindenau M, Funsterer C, Pulst SM, Baser ME et al. Spinal tumors in patients with neurofibromatosis type 2. AJR Am J Roentgenol. 1996;166(5):1231.[Pubmed]
[11].Patronas NJ, Courcoutsakis N, Bromley CM, Katzman GL, MacCollin M, Parry DM. Intramedullary and spinal canal tumors in patients with neurofibromatosis 2: MR imaging findings and correlation with genotype. Radiology 2001; 218:434-42. [Pubmed]
[12].Dow G, Biggs N, Evans G, Gillespie J, Ramsden RT, King A. Spinal tumors in neurofibromatosis type 2: is emerging knowledge of genotype predictive of natural history? J Neurosurg Spine 2005; 2:574 –9. [Pubmed]

